
Introduction to COPD
Why do we need to understand the role of the epithelium and epithelial cytokines in COPD?
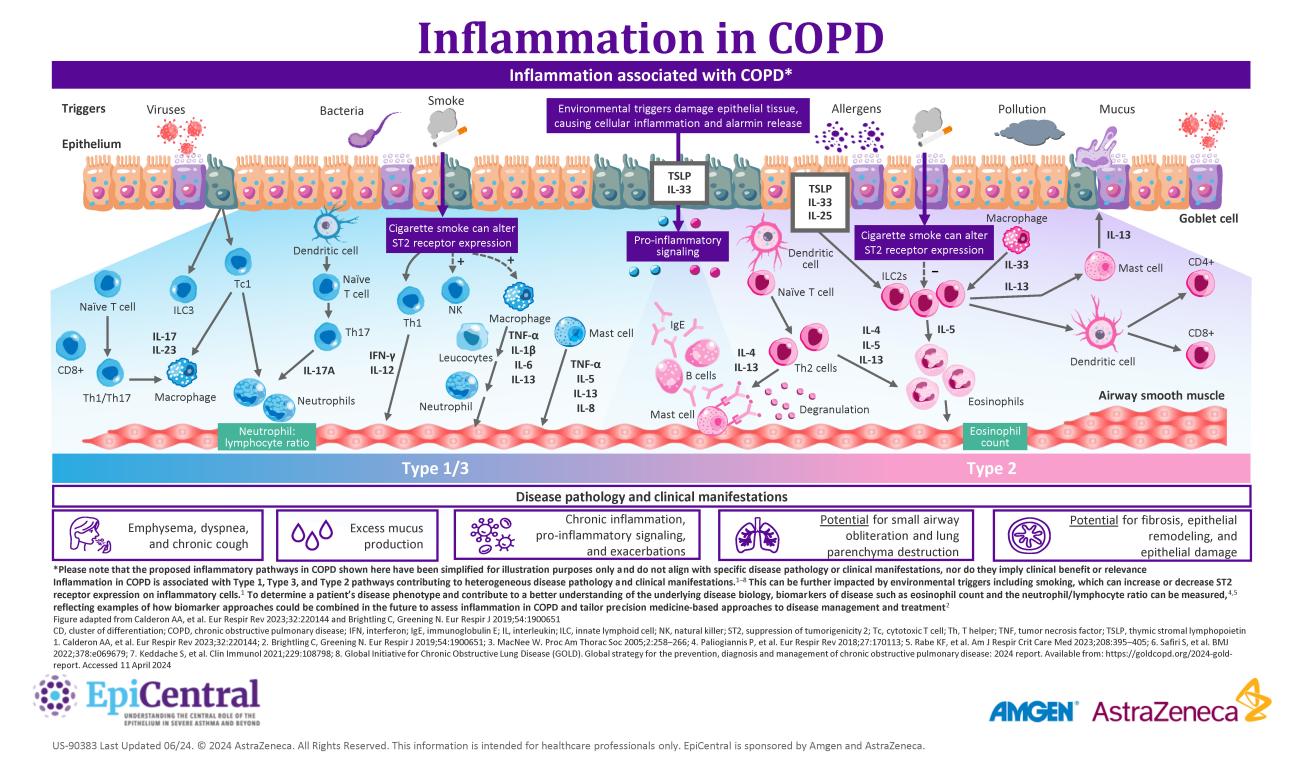
COPD is a complex, heterogeneous, progressive inflammatory lower airway disease,6 which is characterized by exacerbations, inflammation, tissue destruction, and airway remodeling.6 Owing to its complexity, patients with COPD present with a wide variety of disease symptoms, comorbid conditions, and underlying pathophysiology.6,9 The heterogeneity in COPD is driven by different molecular mechanisms (endotypes) and influenced by complex host-environment interactions at the protein-to-cell and tissue-to-organ level.6,8 Together, these interactions determine the clinical presentation of disease characteristics (phenotypes).6,8
The role of the epithelium and epithelial cytokines in COPD
The epithelium and epithelial cytokines play a central role in the pathogenesis of COPD; understanding and characterizing this could help to develop novel phenotyping strategies, identify new biomarkers, and tailor clinical decision making1–5
The epithelium and epithelial cytokines play a central role in the pathogenesis of COPD; understanding and characterizing this could help to develop novel phenotyping strategies, identify new biomarkers, and tailor clinical decision making1–5
Module synopsis
- Chronic obstructive pulmonary disease (COPD) is a growing global health crisis and is one of the top three leading causes of death worldwide.5,6 It is estimated that by 2060, COPD and related conditions will account for over 5.4 million deaths each year globally,5 with costs in the USA alone set to increase to more than $800 billion over the next 20 years5
- Many patients continue to experience exacerbations and symptoms, despite receiving guideline-recommended therapy7
- COPD is a heterogeneous, progressive inflammatory lower airway disease,6 for which the risk factors and biological mechanisms involved in disease initiation and progression are not fully understood6
- The presentation of different phenotypes in COPD is impacted by the heterogeneity of underlying molecular mechanisms of disease, as well as variability in complex host environment interactions8
- The epithelium and epithelial cytokines play a key role in driving inflammation in COPD; understanding and characterizing inflammatory profiles in COPD could aid patient phenotyping, biomarker identification, and clinical decision making1–4
Professor Christopher Brightling's, Professor MeiLan Han's and Professor Klaus Rabe's thoughts on the role of the epithelium and epithelial cytokines in COPD
Insights from our EpiCollaborators

Airway inflammation is implicated in both the pathogenesis and progression of COPD.8 Research characterizing the inflammatory pathways activated in different patients with COPD has revealed different disease phenotypes:8
- Neutrophil-associated Type 1 and Type 3 immunity is the most common phenotype8,10
- Eosinophil-associated Type 2 mediated immunity occurs in a smaller proportion of patients8,10
- Autoimmunity is observed in some patients, particularly those with severe disease8,11
Exacerbations, characterized by an acute worsening of disease symptoms, also vary between patients with COPD.6 Exacerbations can be classified according to the underlying predominant inflammatory profile:6
- A pro-inflammatory endotype (bacteria predominant)
- A T helper (Th) type 2 endotype (eosinophil predominant)
- A Th1 endotype (virus predominant)
- A low-inflammatory profile (pauci inflammatory)
The epithelium is repeatedly exposed to a range of environmental factors that have been shown to play a role in COPD disease pathogenesis, including tobacco smoke, infections, and pollutants.3,6,12 When these triggers come into contact with the epithelium, epithelial cytokines, thymic stromal lymphopoietin (TSLP), interleukin (IL)-33 and IL-25, are released, initiating a cascade of immune responses leading to inflammation and structural changes that contribute to the clinical features of airway disease.1,4,13,14
The burden and unmet needs in COPD
Further research is needed to better diagnose and treat COPDCOPD remains a global health crisis as morbidity and mortality rates of chronic respiratory diseases, of which COPD is a leading contributor, continue to increase.6,8 In the USA, COPD is the leading cause of death among chronic lower respiratory diseases.15 In 2022, chronic lower respiratory diseases were responsible for over 147,000 deaths.16
While COPD is known to be a complex and heterogeneous disease, current classification strategies, diagnostic tests and interventions offer limited options to tailor treatment based on patients’ symptoms or the underlying pathophysiology of the disease.6,9
The current mainstay treatment for patients with COPD is inhaled corticosteroid (ICS) therapy in combination with long-acting inhaled bronchodilators, including long-acting β2 agonists and long-acting muscarinic antagonists;17 however, despite treatment, many patients continue to be symptomatic and experience exacerbations.7 Reflexive treatment of COPD exacerbations with oral corticosteroids or antibiotics (or both) has been the standard treatment approach for patients with COPD for >30 years.6
The current biomarkers typically used in COPD to inform inflammatory endotypes include eosinophil count and fractional exhaled nitric oxide (FeNO), which can be used as predictive biomarkers of ICS responsiveness.4,18 A better understanding of the causative mechanisms of COPD and more sensitive diagnostic measures to detect associated biomarkers could enable the development of personalized prevention and treatment strategies for both stable disease and exacerbations at an earlier stage before further damage occurs.8,19
Pathophysiology: the role of the epithelium in COPD
The respiratory epithelium acts as a physicochemical barrier, which may be impaired by inhalation of airborne environmental triggers, leading to exaggerated inflammatory responses and airway remodeling.3 Epithelial cells provide the first line of defense by providing a barrier function through dense intracellular junctions and mechanical clearance of the airways.20 Therefore, epithelial barrier damage in COPD represents a potential link between triggers at the epithelium and COPD exacerbations or worsening of the disease.3
In COPD, a cycle of persistent, repeated injury and abnormal epithelium repair drives chronic airway inflammation and remodeling,12 which causes histological, cellular and molecular changes in the airways of patients with COPD.21 Chronic oxidative stress and inflammation induce premature epithelial senescence, contributing to impaired epithelial integrity, airway inflammation and remodeling.12 Furthermore, defective antioxidant, antiviral and damage repair mechanisms are thought to further increase susceptibility to airway epithelial dysfunction.12
Mucus hypersecretion and ciliary dysfunction in COPD are associated with deregulated basal cell differentiation and overproduction of the mucin 5AC (MUC5AC) by goblet cells of the bronchial epithelium.12,20 This leads to airway obstruction, which is associated with declining lung function as measured by forced expiratory volume in 1 second (FEV1) and increased risk of exacerbations.20
Recent research outlining the concepts of fixed epithelial damage/activation and epithelial extrusion has further highlighted the potential role of epithelial cells in COPD as possible drivers of disease mechanisms.12,22 Additionally, chronic exposure to cigarette smoke, a key risk factor for COPD, can cause changes in the epithelium, including decreased ciliated epithelial cell number, increased goblet cell number, shortened cilia, and reduced beat frequency.12
Dysregulated epithelial barrier function, and its subsequent contribution to further promote disease, is a central element of COPD, similar to asthma.23 These diseases share several features, including altered apico-basal polarization, disrupted junctional protein expression or localization, as well as reduced basal-to-apical transcytosis of immunoglobulins.23 In contrast, altered basal cell differentiation, reduced numbers of ciliated cells, goblet cell hyperplasia and squamous metaplasia, which are associated with mucus hypersecretion and altered mucociliary clearance of inhaled particles/pathogens, are more typically associated with COPD.23
To learn more about the role of the epithelium in other airway diseases, please view the ‘Role of the epithelium in asthma’ module here, the ‘United airway disease’ module here, and the ‘Chronic rhinosinusitis and the central role of the nasal epithelium’ module here.
Pathophysiology: epithelial cytokines are associated with clinical features of COPD
As well as structural changes, airway damage in COPD causes a release of pro inflammatory mediators and damage-associated molecular patterns, such as IL-33 and TSLP, from epithelial cells.24,25 These epithelial cytokines can play both upstream and downstream roles in regulating immune responses in COPD.25
IL-33
IL-33, an epithelial cytokine associated with the pathogenesis of COPD, exists in two forms, a reduced and oxidized form, which have been shown to activate distinct pathways in COPD.25–27
IL-33 has been shown to promote both Type 1 and Type 2 inflammation in vivo.25 The IL-33 receptor, also known as the suppression of the tumorigenicity 2 (ST2),8 is found on several different cell types including airway endothelial cells, Type 2 innate lymphoid cells (ILC2s), mast cells, myeloid cells, natural killer (NK) cells, Th1 and Th2 cells, cytotoxic T cells (Tcs), NK T cells and basophils.25 Activation of these cells by IL-33 can induce the release of numerous cytokines, including IL-5, IL-13, IL-8 and tumor necrosis factor alpha (TNF-α).25 These cytokines can have a number of downstream inflammatory effects, including inducing immunoglobulin E (IgE) production from B cells and activating eosinophils and mast cells.8 If dysregulated, the marked release of cytokines and the subsequent increase in airway inflammation can compromise the integrity of the airway, resulting in COPD symptoms and an increased exacerbation risk.8
The oxidization of IL-33 has been shown to activate the receptor for advanced glycation end products / epidermal growth factor receptor (RAGE/EGFR) pathway, leading to increased mucus production, airway obstruction, and structural remodeling.27,28
IL-33 expression has been shown to be increased in patients with COPD compared with healthy controls.29,30 In two small cohort studies, IL-33 expression was increased in patients with very severe disease.31,32 In addition, IL-33 expression was also significantly correlated with smoking-pack years.29
TSLP
TSLP, another epithelial cytokine, has also been linked with the pathogenesis of COPD.33 Upon release from the airway epithelium, TSLP can drive a range of Type 2 and non-Type 2 processes with effects on a broad range of cells, including eosinophils, basophils, mast cells, airway smooth muscle cells, ILC2s, dendritic cells, platelets, sensory neurons and macrophages.25
Research has shown that TSLP can directly promote the differentiation and response of Th2 cells in the lung to promote COPD through an independent pathway.34
Studies investigating TSLP expression in patients with COPD have shown higher levels of this epithelial cytokine in disease,35,36 including in induced sputum and broncho-alveolar lavage, compared with controls.33,35 Increased epithelial TSLP mRNA expression has also been identified in nonexacerbating patients with COPD who were exacerbation free for 1 month prior to the study compared with non-smoking controls,35 and another study suggested that increased TSLP expression may be associated with distinct subtypes.37 Serum concentrations of TSLP and IL-17A have also been shown to be higher in patients with COPD than those in healthy controls.34 The cause of elevated TSLP mRNA expression has been associated with moderate-to-severe airflow obstruction and heavy smoking in patients with COPD.35,37
TSLP may drive the pathophysiology in COPD via effects on a variety of downstream cells. For example, airway smooth muscle cells isolated from patients with COPD have been found to show an increase in TSLP protein expression compared with healthy controls.38 Activation of airway smooth muscle cells by TSLP can promote airway remodeling, which is correlated with airflow limitation and decreased lung function in asthma.39
IL-25
Unlike IL-33 and TSLP, the role of IL-25 in the pathophysiology of COPD remains largely unknown. Although IL-25 has been shown to play a role in several inflammatory respiratory conditions,40 the potential link between IL-25 expression and clinical manifestations of COPD requires further research
Evidence for the role of IL-25 in other respiratory diseases suggests that this epithelial cytokine may contribute to inflammation in COPD. For example, IL-25 has been shown to perpetuate the Type 2 inflammatory response through activation of Th2 cells, basophils, eosinophils, and mast cells in other respiratory diseases.41 Additionally, airborne allergens, a key trigger in COPD, have been shown to cause a rapid release of pre-stored IL-25 and an increase in IL-25 mRNA transcription.40 Furthermore, in the airway epithelium, IL-25 induces proallergic chemokine production and cellular changes associated with COPD, including an increase in goblet cells and mucus secretion, epithelial cell hyperplasia and hyperreactivity.40
IL-25 has been shown to be significantly increased in patients with COPD who also have high levels of TSLP, compared with patients with COPD who have low levels of TSLP.36 This could reflect the fact that epithelial-derived IL-25 can function in an autocrine manner, causing the epithelium to produce more IL-25 as well as other cytokines that are known to drive inflammation in COPD, including TSLP.40,42
Pathophysiology: the inflammatory cascade of COPD
The epithelial-derived alarmins IL-33 and TSLP can influence both Type 2 and non-Type-2 inflammation.25 There are two main inflammatory phenotypes in COPD: neutrophilic and eosinophilic inflammation.8
Neutrophil-associated COPD
Neutrophil-associated COPD drives Type 1 and Type 3 inflammation and is the predominant inflammatory phenotype in patients with COPD.4,8,10 Following epithelial cell damage and activation from environmental triggers including cigarette smoke, epithelial cells can secrete factors, such as chemokine CXC ligand 8 (CXCL8), which attract neutrophils via the chemokine CXC receptor 1 (CXCR1) and CXCR2.8,43 Infiltrating neutrophils produce serine proteases and elastolytic enzymes such as matrix metalloproteases (MMP) 8,9 and proteinase-3, which can contribute to the hallmark disease features of COPD including mucus hypersecretion and fibrosis of the small airways.43 Macrophages can also be activated in this process and induce Th17 or group 3 innate lymphoid cells to produce IL‐17, which stimulates the release of IL‐6 and CXCL8 from epithelial cells.44 Despite being the most common phenotype in patients with COPD, currently, there is a lack of specific biomarkers for Type 1/3 phenotypes.4,45
Eosinophil-associated COPD
In contrast, eosinophilic inflammation, which drives Type 2 inflammation, is associated with Th2 cells and ILC2s, which secrete the Type 2 inflammatory cytokines IL-4, IL-5, and IL-13 in response to epithelial-derived IL-33 and TSLP.8 Following this, eosinophils and other Type 2 inflammatory cells that have been activated by IL-4 and IL-13 traffic to the lungs.4 This is induced by the release of chemoattractants, including eotaxin-3 from airway epithelial cells.4
The smoking status of patients with COPD and exposure to cigarette smoke have been shown to impact the inflammatory cascade, altering gene expression and release of inflammatory cytokines and chemokines by bronchial epithelial cells and circulating inflammatory cells.46,47 In particular, active smoking has been shown to reduce small airway intraepithelial eosinophil counts in patients with COPD.46
There is no current evidence to suggest that clinical manifestations are discrete to specific inflammatory profiles.48,49 Instead, there is increasing support for the use of biomarkers that enable a more thorough understanding of the underlying disease biology in different patients to allow for:50
- Accurate identification and stratification of patients who are most likely to benefit from different therapeutics (predictive biomarkers)
- Prediction of individual therapeutic responses to drugs (response biomarkers)
- Segregation of patients based on their likely outcome responses (prognostic biomarkers)
This approach to disease management and treatment, termed precision medicine, aims to tailor treatment approaches to reflect the heterogeneity and complexity of COPD in different patients.49,50
Some of the biomarkers that are currently used for COPD include blood and sputum eosinophil count and FeNO; eosinophil count can be used as predictive biomarkers of ICS responsiveness and have been associated with exacerbation risk.4,9 FeNO levels are associated with eosinophil count; however, studies have found that FeNO can be confounded by the effects of smoking.4,9 Additionally, data on the real-world application of these biomarkers within different patient populations to predict features of exacerbations in COPD have been inconsistent, suggesting the need for new biomarkers for COPD.4,9,18
Identifying new biomarkers is an active area of research in COPD as this could enable earlier detection and diagnosis of COPD at a stage before further damage occurs and may enable the detection of the disease phenotype and the identification of personalized prevention and treatment strategies for both stable disease and exacerbations.8,9,19
Examples of new biomarkers in COPD may include:
- The neutrophil-lymphocyte ratio (NLR) has been investigated as a biomarker of systemic inflammation, reflecting the recruitment of both the main white blood cell populations (lymphocytes and neutrophils) in COPD.18 NLR can be investigated using samples from routine blood analysis and has been suggested as a biomarker of both disease severity and prognosis18
- Exhaled breath sampling, termed breathomics, is emerging as an attractive choice to detect biomarker signatures of disease owing to its non-invasive nature and potential applications in diagnosis, disease monitoring,51 and the identification of clinically treatable traits, such as airway inflammation,8,52 eosinophilia, and risk of infection/exacerbation.53 For example, increasing evidence supports a role for FeNO as a biomarker that can be sampled from a simple breath test, which is correlated with activated Type 2 inflammation and sputum eosinophil count in patients with COPD.4,9 Indeed, breathomics could be used in predicting the inception of COPD, as well as inflammatory phenotyping, exacerbation prediction, and treatment stratification in other diseases53
- Transcriptome profiling is also being explored to identify gene expression signatures associated with COPD, such as those related to treatment response and lung function decline that can be identified from bronchial epithelial brushes, biopsies, lung tissue or peripheral blood2
As well as identifying new biomarkers, researchers are also exploring sites of the body for non-invasive biomarker detection, including detection at the nasal epithelium,2 for which a gene expression profile has been detected that can distinguish between patients with and without severe COPD.54 Further research investigating the role of the epithelium, and epithelial cytokines, within COPD may enable the identification of novel biomarkers, phenotyping strategies or targeted treatments.1–4
The clinical importance of understanding pathobiology and phenotypes in COPD
COPD is a highly complex, heterogeneous disease, and the features of COPD need to be reflected in interventions as part of a holistic approach to treatment.6 Improving understanding of the pathobiology of COPD, including the inflammatory pathways that result in the presentation of different disease symptoms, has been recognized as a priority to enable disease phenotyping and development of tailored treatments to improve patient care.2,6 Advancing knowledge of the role of the epithelium and epithelial cytokines may further clarify some of the aspects of COPD biology that are not fully understood, including:
- How does COPD start? What are the early triggers?
- How do we identify people at risk earlier to avoid disease progression?
- What is the underlying biology in COPD responsible for the different phenotypes, and how can this be linked to novel treatment targets?
The content for this module was created with the support of Professor Christopher Brightling, Professor MeiLan Han, and Professor Klaus Rabe.
RELATED RESOURCES
Like this page? We have a host of other resources available in the ‘Scientific and Resource Library’ where you can find out more.
References
1. Roan F, et al. J Clin Invest. 2019;129:1441–1451; 2. van den Berge M, Faiz A. Am J Respir Crit Care Med. 2022;205:139–140; 3. Aghapour M, et al. Eur Respir Rev. 2022;31:210112; 4. Rabe KF, et al. Am J Respir Crit Care Med. 2023;208:395–405; 5. Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the prevention, diagnosis and management of COPD: 2024 report. Available from: https://goldcopd.org/2024-gold-report/. Accessed April 11 2024; 6. Stolz D, et al. Lancet. 2022;400:921–972; 7. Sansbury LB, et al. Int J Chron Obstruct Pulmon Dis. 2022;17:415–426; 8. Brightling C, Greening N. Eur Respir J. 2019;54:1900651; 9. Flynn C, Brightling C. Respirology. 2023;28:421–422; 10. Keddache S, et al. Clin Immunol. 2021;229:108798; 11. Wen L, et al. Front Immunol. 2018;9:66; 12. Raby KL, et al. Front Immunol. 2023;14:1201658; 13. McBrien CN, Menzies-Gow A. Front Med (Lausanne). 2017;4:93; 14. Varricchi G, et al. Front Immunol. 2018;9:1595; 15. CDC Morbidity and Mortality Weekly Report (MMWR). Chronic Obstructive Pulmonary Disease Mortality by Industry and Occupation — United States, 2020. Available from: https://www.cdc.gov/mmwr/volumes/71/wr/mm7149a3.htm. Accessed May 29 2024; 16. CDC National Center for Health Statistics. Leading Causes of Death. 2024. Available from: https://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm. Accessed May 29 2024; 17. Quint JK, et al. NPJ Prim Care Respir Med. 2023;33:27; 18. Paliogiannis P, et al. Eur Respir Rev. 2018;27:170113; 19. Franssen FM, et al. Int J Chron Obstruct Pulmon Dis. 2019;14:1465–1484; 20. Kotlyarov S. Int J Mol Sci. 2022;23:985; 21. Lange P, et al. Respirology. 2021;26:298–321; 22. Thomas B, et al. COPD. 2021;18:657–663; 23. Carlier FM, et al. Front Physiol. 2021;12:691227; 24. Furci F, et al. Biomedicines. 2023;11:437; 25. Calderon AA, et al. Eur Respir Rev. 2023;32:220144; 26. Takatori H, et al. Front Immunol. 2018;9:2004; 27. Strickson S, et al. Eur Respir J. 2023;62:2202210; 28. Vallath S, et al. Eur Respir J. 2014;44:513–522; 29. Joo H, et al. BMC Pulm Med. 2021;21:86; 30. Zhou Y, et al. J Transl Med. 2023;21:902; 31. Byers DE, et al. J Clin Invest. 2013;123:3967–3982; 32. Kearley J, et al. Immunity. 2015;42:566–579; 33. Anzalone G, et al. Exp Mol Med. 2018;50:1–12; 34. Liu M, et al. Front Pharmacol. 2021;12:695957; 35. Ying S, et al. J Immunol. 2008;181:2790–2798; 36. Wu L, et al. Int J Clin Exp Med. 2019;12:4942–4948; 37. Yamada H, et al. COPD. 2020;17:59–64; 38. Zhang K, et al. Am J Physiol Lung Cell Mol Physiol. 2007;293:L375–L382; 39. Redhu NS, et al. Sci Rep. 2013;3:2301; 40. Borowczyk J, et al. J Allergy Clin Immunol. 2021;148:40–52; 41. Chan R, et al. J Allergy Clin Immunol Pract. 2022;10:1497–1505; 42. Duchesne M, et al. Front Immunol. 2022;13:975914; 43. Butler A, et al. COPD. 2018;15:392–404; 44. Barnes PJ. Allergy. 2019;74:1249–1256; 45. Thulborn SJ, et al. Respir Res. 2019;20:170; 46. Beech A, et al. ERJ Open Res. 2024;10:00870–02023; 47. Chung KF. Eur Respir J Suppl. 2001;34:50s–59s; 48. O’Donnell R, et al. Thorax. 2006;61:448–454; 49. Sidhaye VK, et al. Eur Respir Rev. 2018;27:180022; 50. Leung JM, et al. Eur Respir J. 2019;53:1802460; 51. Kiss H, et al. Micromachines (Basel). 2023;14:391; 52. Borrill ZL, et al. Eur Respir J. 2008;32:472–486; 53. Bos LD, et al. J Allergy Clin Immunol. 2016;138:970–976; 54. Boudewijn IM, et al. Respir Res. 2017;18:213.